
Hao Zhou (周浩)
Research Associate Professor
Insititute for AI Industry Research (AIR), Tsinghua
University
![]() Email: zhouhao@air.tsinghua.edu.cn & haozhou0806@gmail.com
Email: zhouhao@air.tsinghua.edu.cn & haozhou0806@gmail.com






Hello! I am a Research Associate Professor at the Institute for AI Industry Research (AIR), Tsinghua University. My work centers on building Generative AI for discrete symbols (text and proteins). I focus on Large Language Models (LLMs) and its applications to scientific discovery, with special interests in biological agents. Previously, I was a Research Scientist/Manager at Bytedance, where I led the ByteDance AI Lab research teams on text generation and AI drug design. Here is my bio in Chinese 中文 and my CV.
- I lead the GenSI research group at Tsinghua AIR
- I co-lead the SIA Lab, the joint laboratory of THU-AIR and Bytedance Seed on cutting-edge LLM research.
- Professional Service: Area Chair of EMNLP 2022; ACL 2023; NeurIPS 2022–2025; ICML 2023–2025; ICLR 2023–2026.
Recent Highlight
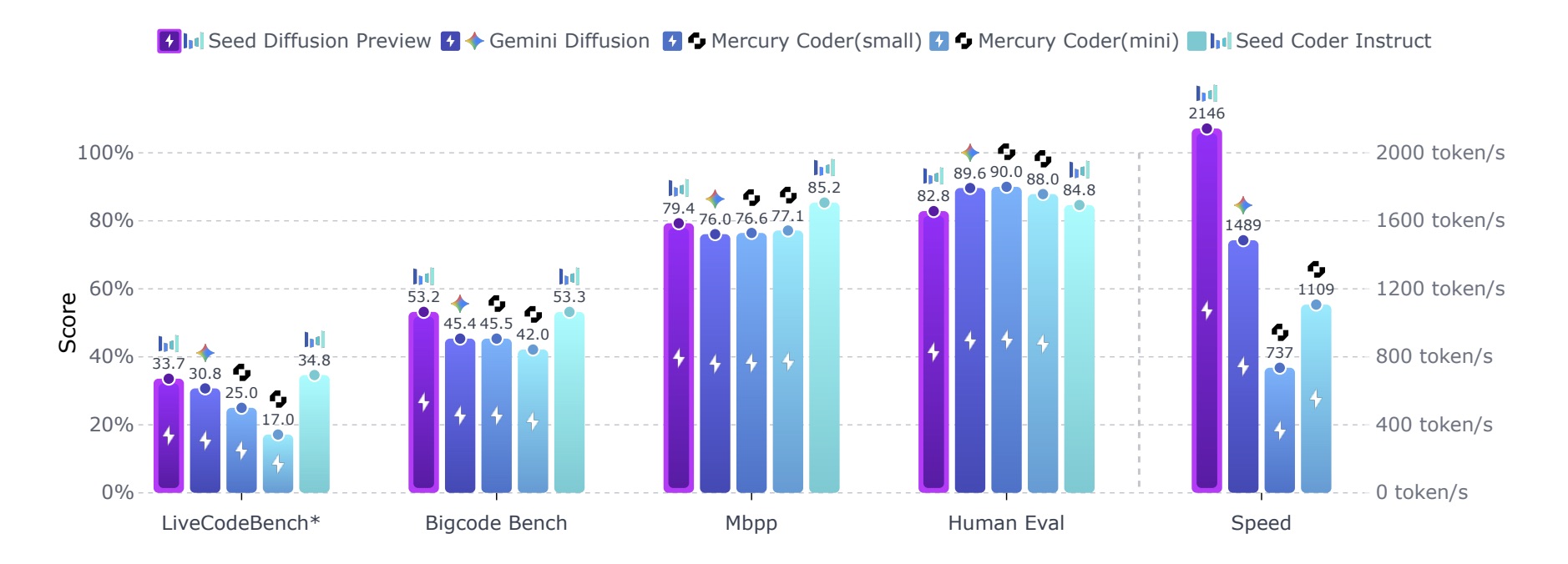
Seed Diffusion Preview
A large scale language model based on discrete-state diffusion, achieves an inference speed of 2,146 token/s, outperforming its Google Gemini Diffusion and Mercury Counterparts.
🚀 全面超越google的Gemini-diffusion,一秒钟生成2000+的词,性能不掉!
Page Paper Demo CitationsNEW
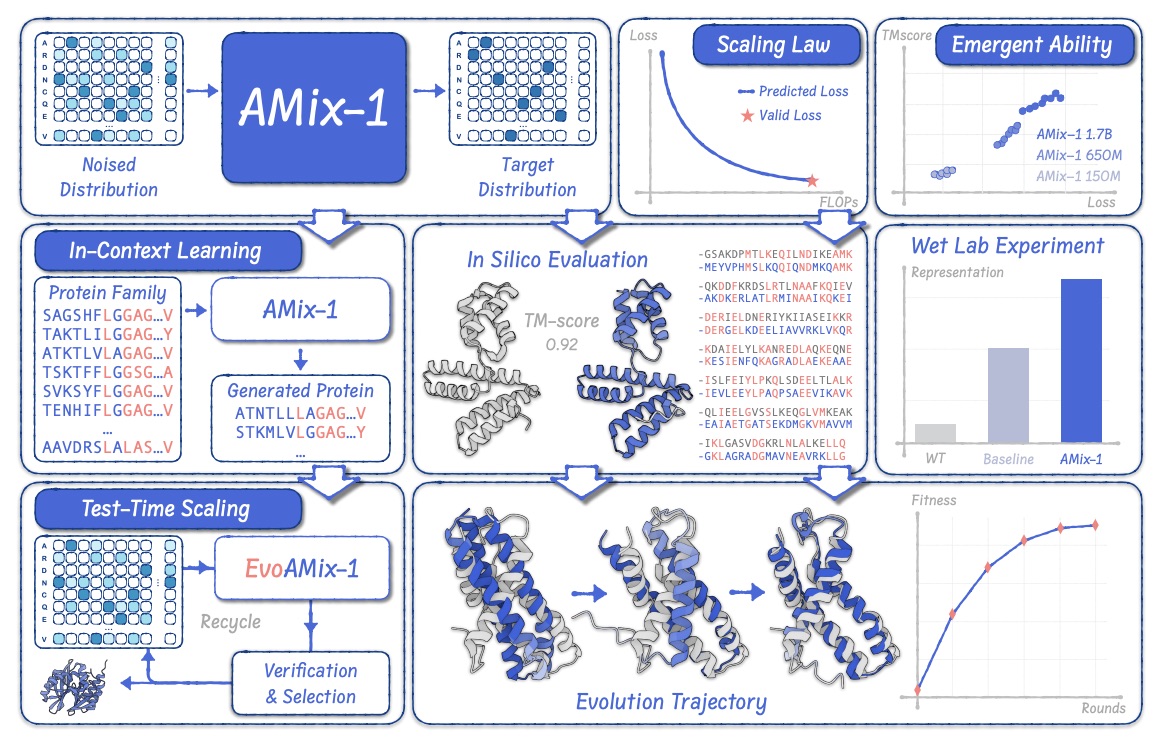
AMix-1: A Pathway to Test-Time Scalable Protein Foundation Model
We propose a systematic paradigm, including scaling laws, structural emergent ability, in-context learning, and test-time scaling, for crafting protein foundation models, emphasizing a road map towards scalable protein design. We design a high-activity AmeR variant with a 50× improvement over the wild type.
全新可推理、可Test-time-Scaling的蛋白质基座 AMix-1🔥!
我们提出了AMix-1,首次尝试以Scaling Law、Emergent Ability、In-Context Learning 和 Test-time Scaling
的系统化方法论来构建蛋白质基座模型,并和清华张数一老师团队合作在AmeR蛋白设计上提升其能力50倍!
Page Paper CodeNEW Model CitationsNEW
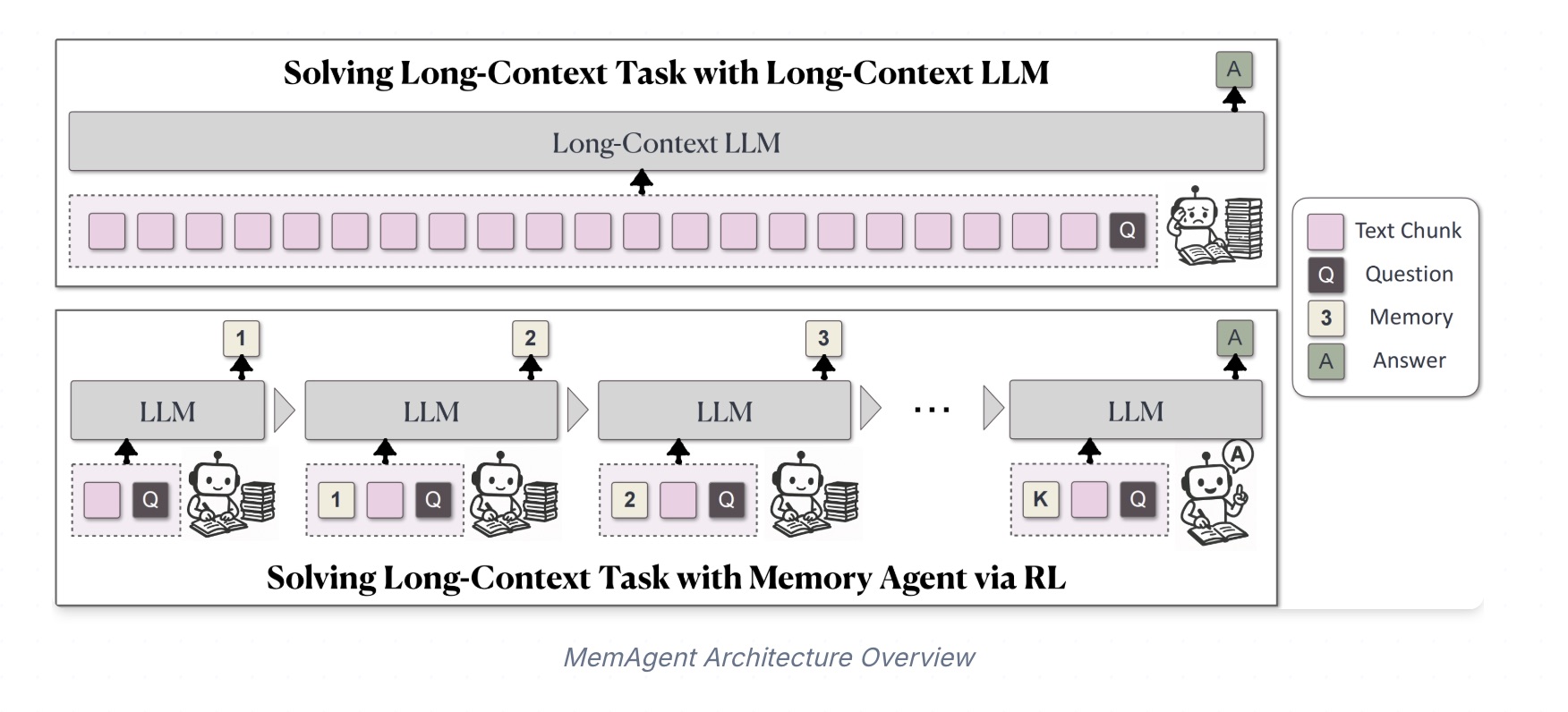
MemAgent: Reshaping Long-Context LLM with Multi-Conv RL based Memory Agent
MemAgent has demonstrated superb long-context capabilities, being able to extrapolate from an 8K context trained on 32K text to a 3.5M QA task with performance loss < 5% and achieves 95%+ accuracy in 512K RULER test.
🔥多轮对话RL + LLM外挂Notepad 🗒️ = Memory Agent.
🚀不需要改动任何网络架构(线性注意力/稀疏注意力),一切交给RL,LLM能自己学到在notepad里保留哪些关键信息并不断更新. 使用具有多轮独立对话的Agent处理长文并用DAPO算法进行了端到端训练。
🏆仅使用8K Context的14B模型可从训练时的32K长度无损外推至3.5M,并且在512K长度的RULER测试集中取得95%+的成绩,7B模型也在测试中超越了更大尺寸的专门长文模型.
Page Paper CodeNEW Data Model CitationsNEW
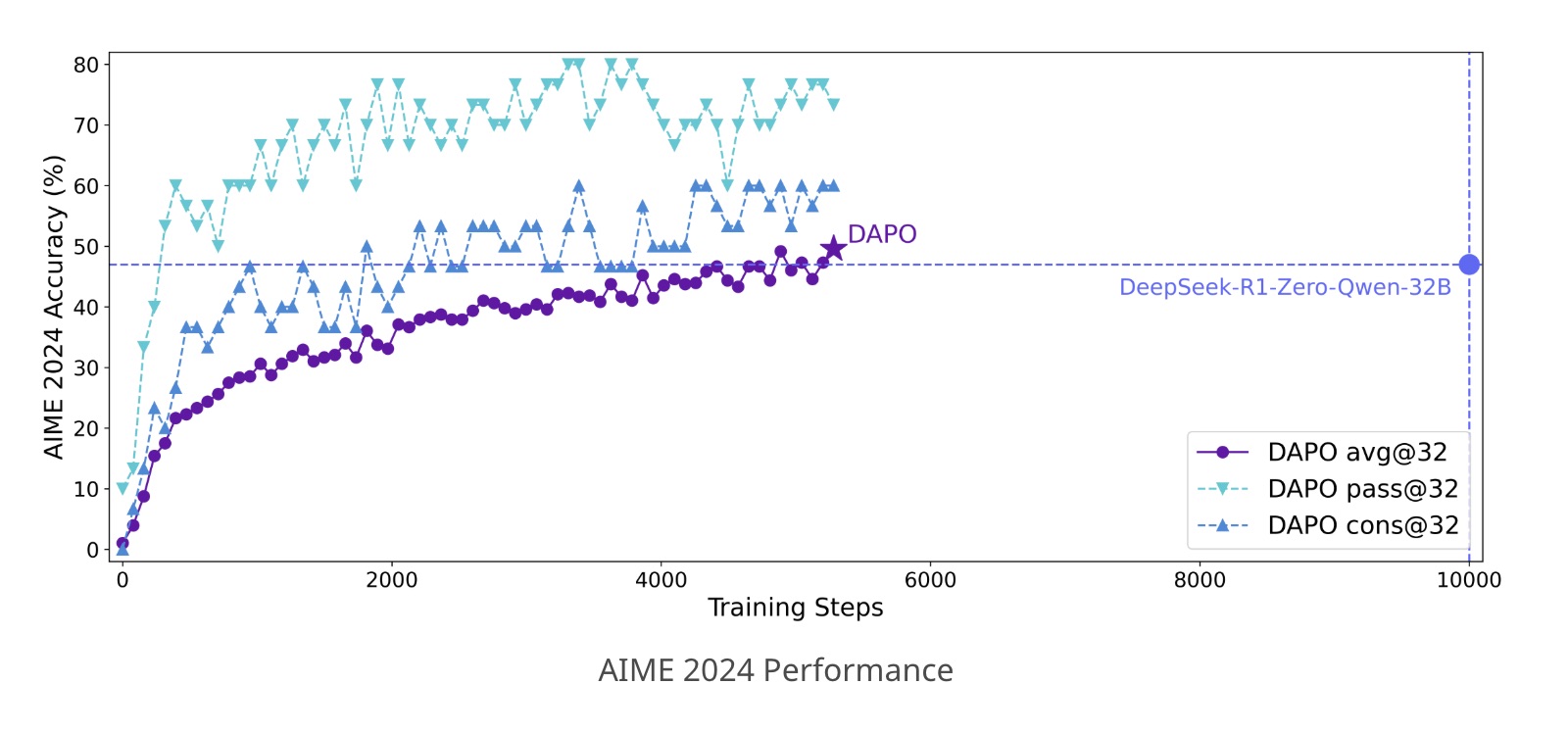
DAPO: an Open-Source LLM Reinforcement Learning System at Scale
We propose the Decoupled Clip and Dynamic sAmpling Policy Optimization (DAPO) algorithm. By making our work publicly available, we provide the broader research community and society with practical access to scalable reinforcement learning, enabling all to benefit from these advancements. Our system is based on the awesome verl framework. Thanks for their great work! Applying DAPO training to Qwen2.5-32B base model proves to outperform the previous state-of-the-art DeepSeek-R1-Zero-Qwen-32B on AIME 2024, achieving 50% accuracy with 50% less training steps.
🚀我们提出了DAPO,一个工业级别的,训练稳定高效率的,框架、数据、训练策略完全开源的大规模强化学习系统,RL from Pretrained (Qwen2.5-32B) 在 AIME 上跑到了50分,超越了之前 DeepSeek 在 R1 paper 里达到的的47分SOTA,欢迎试用!
Page Paper CodeNEW Data Model CitationsNEW